
NGS Data Analysis Bottlenecks
One of the biggest bottlenecks in next generation sequencing (NGS) today is data analysis, which is surprising, considering that we have massive compute available at our fingertips, the available infrastructure to process thousands of samples in parallel, and plenty of programming libraries that simplify complex statistical operations into something resembling plain English.
So why do so many of us get stuck analyzing NGS data for days, even weeks? When Basepair founder Amit Sinha, an instructor at Harvard Medical School, began work on the web platform, he looked back at his decade of computational biology experience – and his colleagues’ most persisting holdups – to spotlight any unnecessary congestion in the NGS data analysis process.
The problem, Amit discovered, wasn’t the lack of technical resources, but a disjointed approach that required multiple pieces of command line software, file conversion between programs, and wasted time waiting for various algorithms to finish. Further, the results were spread across dozens of files, and information of interest had to be forcefully dug out.
Faster alternatives required a bioinformatician to dedicate precious time working in a CLI to run automated scripts, getting IT to help out in parallelizing workflows if dozens or hundreds of samples were involved, and generally leaning on team members with overflowing schedules. This wasn’t ideal either.
Having experienced every bottleneck imaginable in the NGS data analysis process, Amit created Basepair as a one-stop-shop that takes raw fastq files and automatically performs complex analytics operations based on your chosen pipeline. The result is a ready report and a series of interactive graphs that allow researchers and bioinformaticians to move on to downstream analysis without getting stuck juggling algorithms and waiting for schedules to clear up.
Since multiple analyses in Basepair run in parallel, and all compute is hosted on powerful virtual machines in the cloud, the days and weeks traditionally imparted on analyzing multiple samples is reduced to under an hour.
The bread and butter of the Basepair web platform is automated workflows, which have saved teams at Harvard Medical School, Stanford, Memorial Sloan Kettering Cancer Center, and other world-class institutions thousands of hours.
Automated Workflows
At Basepair, we take several steps to ensure end user scientists get fast, accurate results with as few clicks – and as little hands-on time – as possible. We have automated the most useful workflows as ready-to-use pipelines for your NGS research, with several workflows available for each sequencing method.
For your RNA-Seq, DNA-Seq, ChIP-Seq, or ATAC-Seq data we feature custom pipelines modeled after the latest research in bioinformatics; our own bioinformaticians have even tweaked algorithms in these pipelines to ensure sequence data is interpreted with the highest accuracy.
Where the more expert user is concerned, we’ve heard the war stories from our fellow bioinformaticians and lab directors: waking up bleary-eyed in the middle of the night to check on the results of an NGS data analysis pipeline, converting files to different formats, setting up the next step of their downstream analysis, and groggily shifting back to bed! Having gone through the same inane hassle, at Basepair we also understand the utility of a robust API that can automate manual tasks and will touch on that later.
For now, let’s explore a couple options available for RNA-Seq to get a sense of how automated workflows make life easier for both individual bench scientists and research teams of any size.
RNA-Seq Workflows
RNA-Seq is most commonly used for differential gene expression analysis, as well as detecting fusion events, genetic variation, novel transcripts, and a host of other biological phenomena.
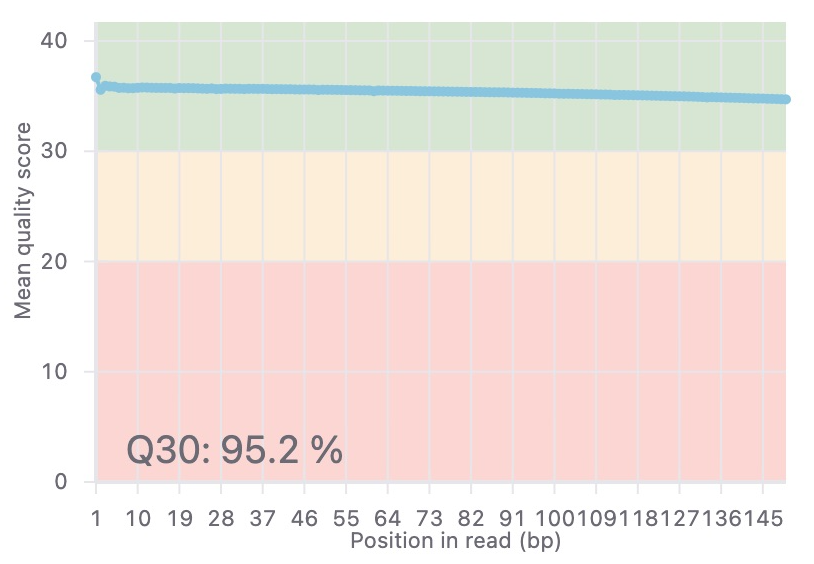
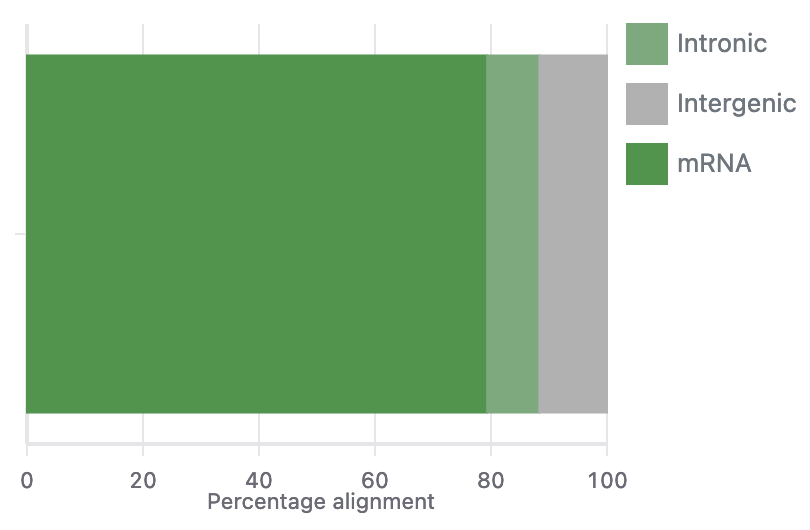
After uploading your data to Basepair, our Alignment/Expression count pipeline can be started with just a few clicks. This pipeline will automatically check the quality of your raw sequencing data, remove sequence contaminants if they are present, and align the data to your genome-of-interest using STAR. Raw and normalized gene expression levels are then quantified using featureCounts, and you can easily view your data with our built-in IGV browser. Whether you have six samples or 6,000 samples, Basepair allows them to be processed in parallel on Amazon’s cloud resources to quickly provide you with results.
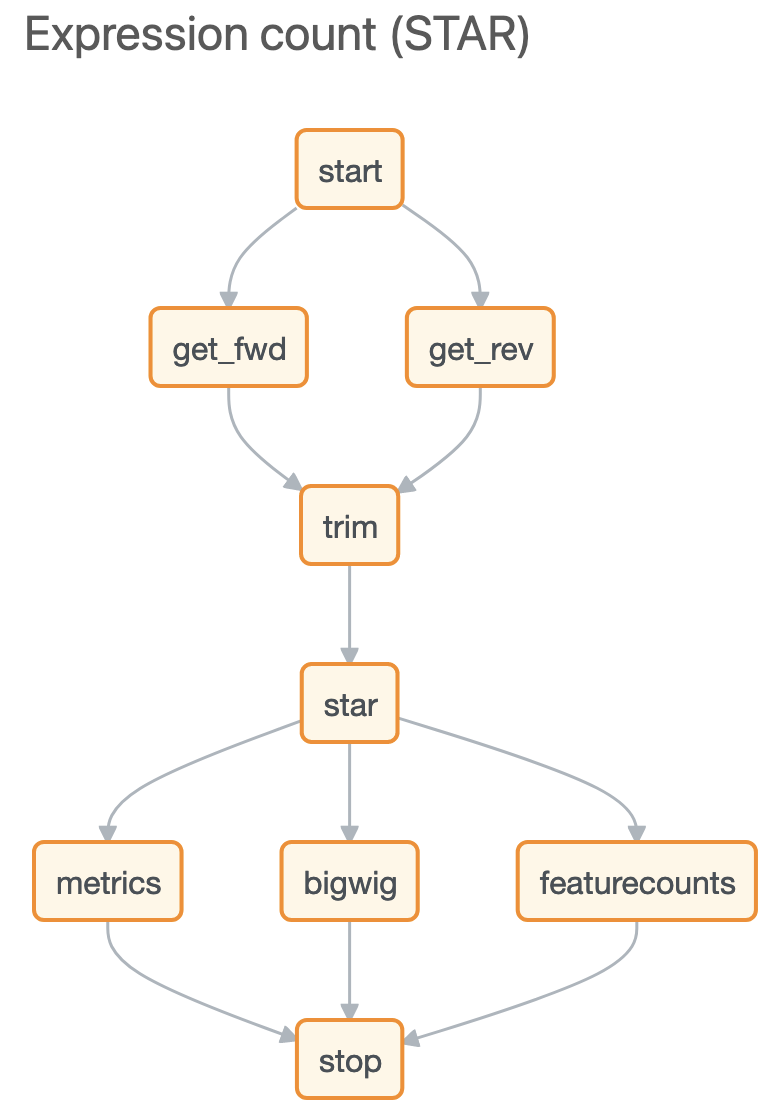
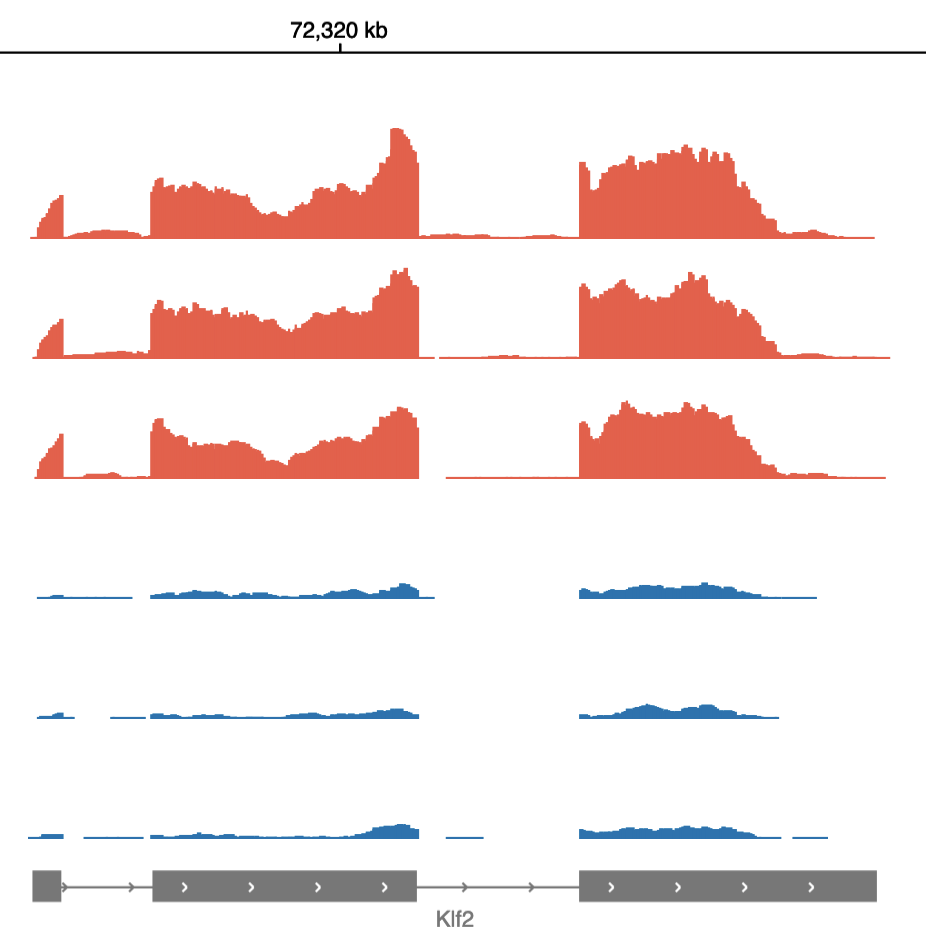
After aligning your data, Basepair’s differential expression analysis pipeline will automatically identify up- and down-regulated genes using DESeq2, perform Principle Component Analysis (PCA), as well as gene ontology (GO) and pathway enrichment analyses using GSEA. The results of these analyses are displayed using interactive plots and tables that allow you to quickly explore your data.
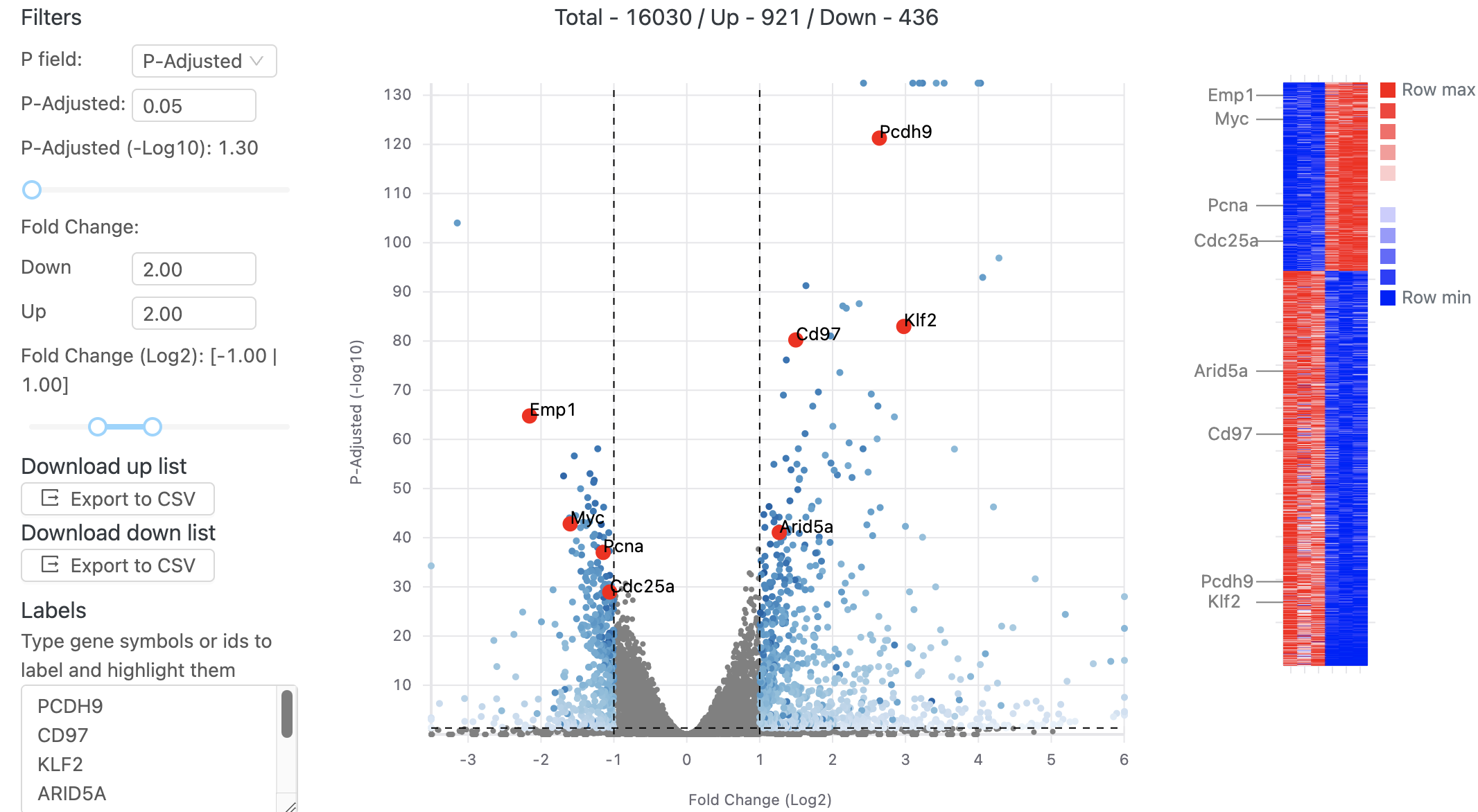
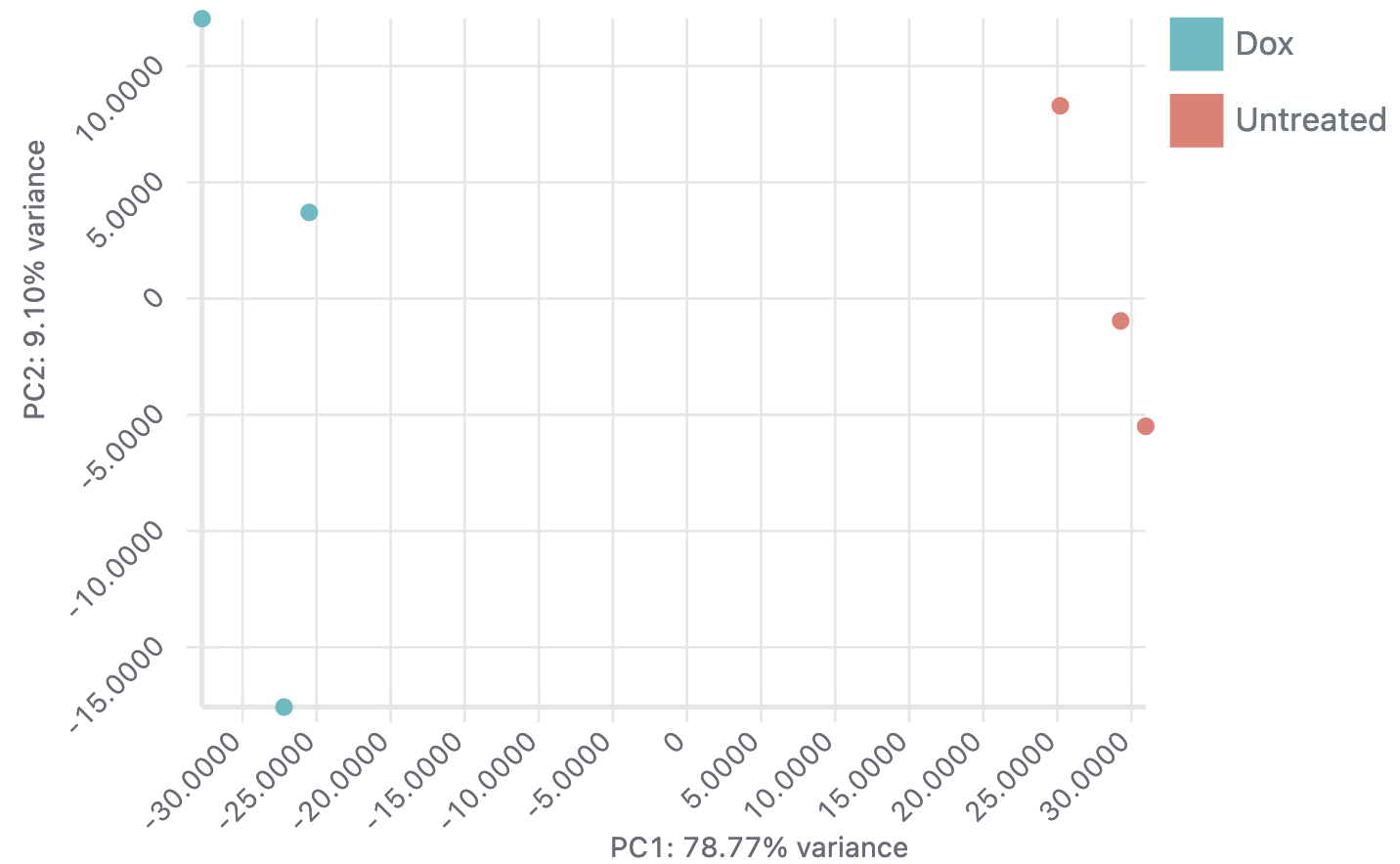
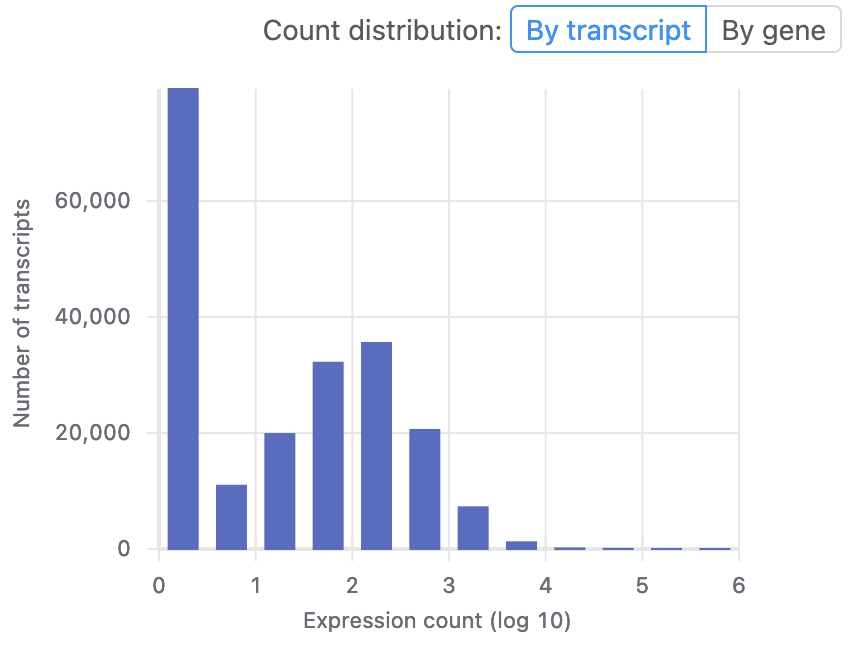
For more in-depth transcriptomic analyses, we also provide pipelines that assess splicing with tools such as cufflinks/cuffdiff, leafcutter, and variant calling using GATK.
APIs
We’ve put together a powerful Application Programming Interface (API) for Python and command line, and made them as readable, simple, and intuitive as we could. As mentioned previously, Basepair’s graphical user interface is already streamlined for one-click, rapid-fire analysis, but when it comes to dozens, hundreds, even thousands of samples, nothing beats the addition of a solid API.
Check out this additional blog post where we go over a few examples of how our Python API helps automate NGS data analysis at scale. If you’d like to check out the command line API, we’ve written up another helpful post here. (To view our help documentation, you need to be signed in – if you don’t have an account, you can sign up for a free two-week trial in less than a minute using the button at the end of this post.)
Even if you’re not a bioinformatician, or don’t have developer experience, we recommend simply glancing at the examples in that post to get a sense of what you can do with Basepair’s API. Basepair can also help you set up the API and any integrations – you can contact us here for more information.
Basepair has over 50 workflows and counting. Our team is diving deep into the latest research in order to provide you with bioinformatics analyses that are both easy to run and easy to interpret. We made Basepair’s API as readable, simple, and robust as we could. A great API can also help teams scale up to thousands of samples in very little time. Thanks to powerful parallel processing, we’re able to run thousands of analyses simultaneously, accumulating no time debt for additional samples.
Try our full platform immediately free by signing up here, and explore our automated workflows with your own NGS data.