
The performance of NGS alignment tools has continuously improved, but sorting the aligned data now takes more time than alignment. A big leap in sorting was recently initiated with SAMtools version 1.x, with support for parallel processing for sorting.
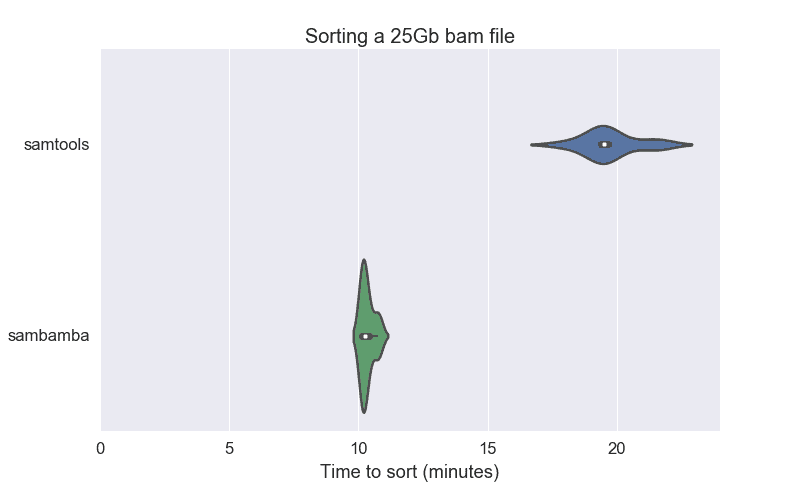
We compared the sorting speed of a 25Gb unsorted BAM file with SAMtools and sambamba. Our results show that sambamba was 2x faster than SAMtools. The following violin plot shows that SAMtools took 20 minutes while sambamba could sort the same file in 10 minutes. The narrow plot for sambamba indicates that its performance is more predictable than SAMtools.
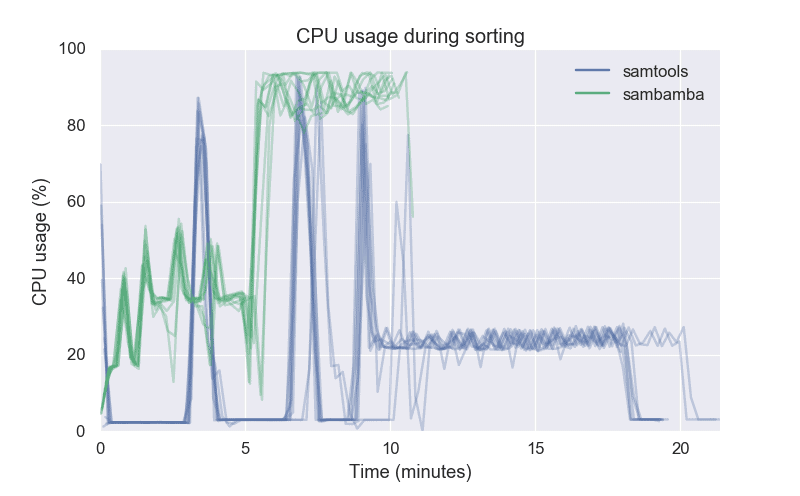
Despite supporting multiple threads, SAMtools is not very good at parallelization. For the first half, SAMtools was using just a single thread with an occasional spike in CPU usage. The CPU usage was little over 20% for the second half. Sambamba used 30-40% CPU for the first half, and then over 90% for the second half.
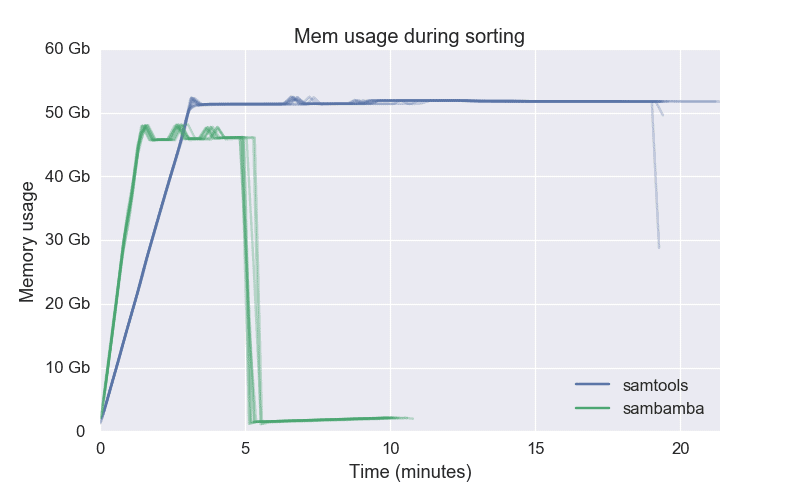
Methods
The tests were run on AWS instance c3.8xlarge (32 cores, 60 Gb RAM) and the files were stored in local storage. The unsorted BAM file was generated by STAR.
SAMtools (version 1.2) and sambamba (version 0.6.3) were run 10 times each, alternatively, to reduce any bias. The applications were run with the following options:
sambamba sort -t 30 -m 45G -o Input.hg19.sambamba-sort.bam Input.hg19.Aligned.out.bam samtools sort -@ 30 -m 1500M -T __sam_tmp__ -o Input.hg19.samtools-sort.bam Input.hg19.Aligned.out.bam
The CPU, memory and disk usage were collected using dstat at 15 second intervals. The plots were generated using Python packages seaborn or matplotlib.